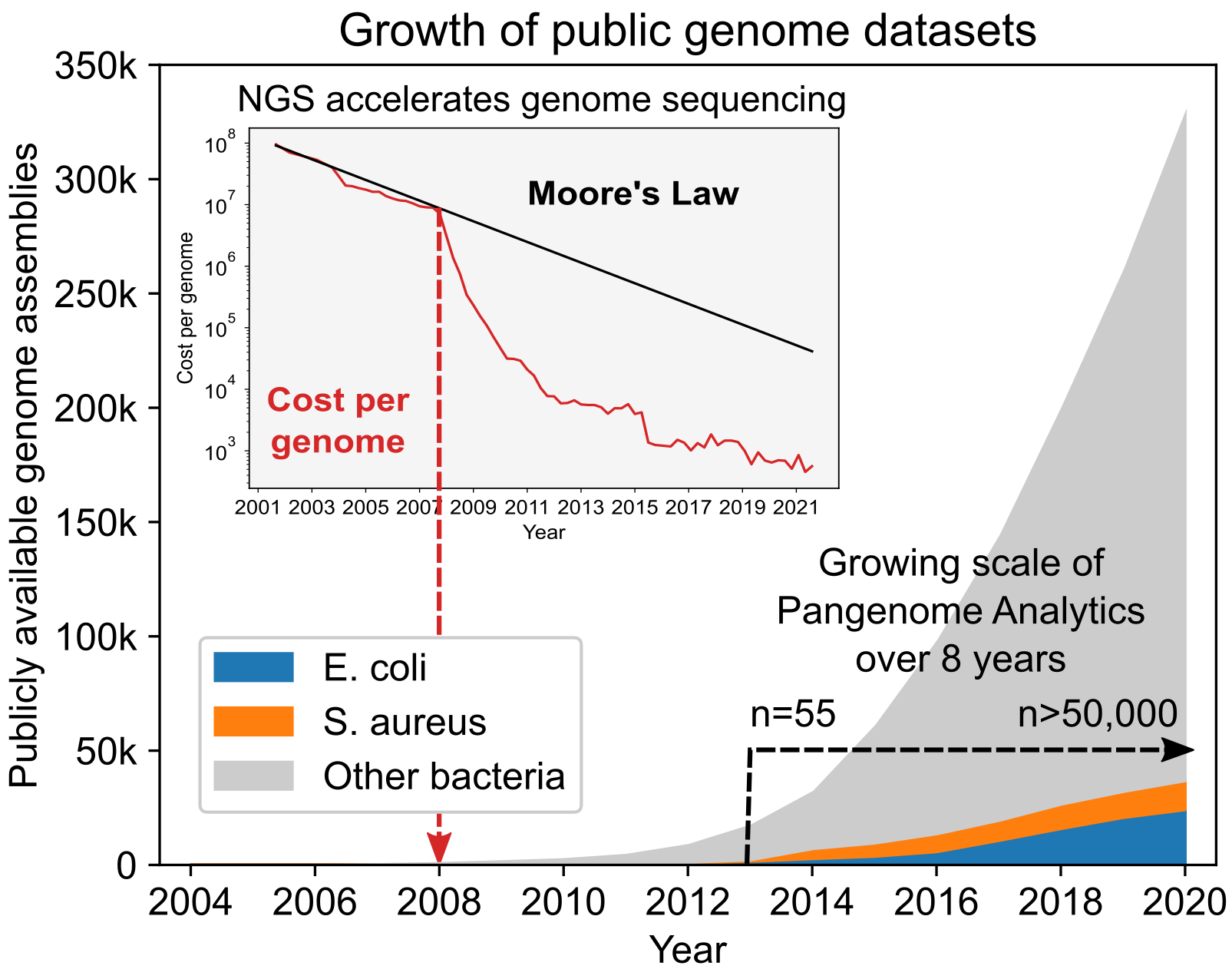
The rapid drop in sequencing costs in the late 2000s led to an exponential increase in the number of WGSs, starting around 2010. We published our first knowledge-enriched pangenome analysis paper in 2013 [8], using 55 WGSs for metabolic reconstructions and GEM formulation. Now, eight years later, we are analyzing >50,000 WGSs from the Web of Life [9] representing 183 bacterial species. The scale of pangenome analysis has thus gone through a three-log increase in eight years; from a few dozen sequences to a comprehensive coverage of publicly available sequences, demonstrating the scalability of our approach.
The totality of genes found in sequenced strains of a species is called the pangenome. The ‘core’ genome is defined as the genes shared by all the strains in a species, while the ‘accessory’ genome is composed of genes that are present in a subset of strains. The growth of the pangenome with the addition of new WGSs can be used to classify a species as having an “open” or “closed” pangenome [1].
Genes in the pangenome can be classified into functional categories (e.g., metabolism, transcription, virulence, etc.). Genes in these categories can next be linked to known proteins and their functions. For metabolism, genes are linked to their encoded enzymes which are then linked to reactions they catalyze. These reactions can be assembled into a reconstructed metabolic network at the genome-scale [2] [3] [4].
Network reconstructions can be converted into genome-scale models (GEMs) that can be computationally compared between strains of the bacterial species under study to delineate their metabolic differences [5] [6] [7] [8].
Over the past decade, the generation rate of omics data sets for pathogens has increased exponentially. Whole genome sequencing (WGS) samples have grown rapidly, doubling approximately every 1.7 years, creating a need for new methods to analyze this data deluge. To address this need, the UC San Diego Systems Biology Research Group (SBRG) has developed state of the art pan-genome analytics methods that can be applied to large datasets to learn lessons at increasing scales:
Single genomes (100): Initial studies of WGSs showed how metabolic networks could be reconstructed, and GEM formulation led to phenotypic predictions [10] [11].
Tens of genomes (101): As the number of WGSs grew, comparative analysis based on presence/absence calls of genes in genomes led to new predictions, such as colonization sites and auxotrophies [8].
Hundreds of genomes (102): At this scale, the prediction of mechanisms underlying infectious potential [12], host range [5] [6] [7], serotypes [13], pangenome properties [5], the virulome [7], and genome organization of virulence factor operons [14] appeared.
Thousands of genomes (103): At this scale, the development of alleleomics [6] emerged, based on the presence/absence calls of a particular allele of a gene in a strain. Allelomics enables the identification of genes under strong selection pressure and their association with antibiotic resistance [15] [16].
Tens of thousands of genomes (104): At this scale, comparative pangenomics between species becomes possible, enabling the identification of various genes that are conserved across branches of the bacterial phylogenetic tree, as well as the discovery of species-agnostic mutation-enriched genes. Within these genes, individual domains which are hypermutable can be identified, as well as conserved regions. Interestingly, 173 genes defined the ‘super-core’ genome of 12 pathogenic species [15] [17] [18].
Citations/Further Reading
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc Natl Acad Sci U S A. 2005;102: 13950–13955.
- Feist AM, Herrgård MJ, Thiele I, Reed JL, Palsson BØ. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7: 129–143.
- Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5: 93–121.
- Palsson BØ. Systems Biology: Constraint-based Reconstruction and Analysis. Cambridge University Press; 2015.
- Seif Y, Kavvas E, Lachance J-C, Yurkovich JT, Nuccio S-P, Fang X, et al. Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits. Nat Commun. 2018;9: 3771.
- Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z, et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat Biotechnol. 2017;35: 904–908.
- Bosi E, Monk JM, Aziz RK, Fondi M, Nizet V, Palsson BØ. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci U S A. 2016;113: E3801–9.
- Monk JM, Charusanti P, Aziz RK, Lerman JA, Premyodhin N, Orth JD, et al. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci U S A. 2013;110: 20338–20343.
- Zhu Q, Mai U, Pfeiffer W, Janssen S, Asnicar F, Sanders JG, et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat Commun. 2019;10: 5477.
- Reed JL, Patel TR, Chen KH, Joyce AR, Applebee MK, Herring CD, et al. Systems approach to refining genome annotation. Proc Natl Acad Sci U S A. 2006;103: 17480–17484.
- Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (i JR904 GSM/GPR). Genome Biol. 2003;4: R54.
- Fang X, Monk JM, Mih N, Du B, Sastry AV, Kavvas E, et al. Escherichia coli B2 strains prevalent in inflammatory bowel disease patients have distinct metabolic capabilities that enable colonization of intestinal mucosa. BMC Syst Biol. 2018;12: 66.
- Seif Y, Monk JM, Machado H, Kavvas E, Palsson BO. Systems Biology and Pangenome of Salmonella O-Antigens. MBio. 2019;10. doi:10.1128/mBio.01247-19
- Choudhary KS, Mih N, Monk J, Kavvas E, Yurkovich JT, Sakoulas G, et al. The Staphylococcus aureus Two-Component System AgrAC Displays Four Distinct Genomic Arrangements That Delineate Genomic Virulence Factor Signatures. Front Microbiol. 2018;9: 1082.
- Kavvas ES, Yang L, Monk JM, Heckmann D, Palsson BO. A biochemically-interpretable machine learning classifier for microbial GWAS. Nat Commun. 2020;11: 2580.
- Kavvas ES, Catoiu E, Mih N, Yurkovich JT, Seif Y, Dillon N, et al. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat Commun. 2018;9: 4306.
- Yang JH, Bhargava P, McCloskey D, Mao N, Palsson BO, Collins JJ. Antibiotic-Induced Changes to the Host Metabolic Environment Inhibit Drug Efficacy and Alter Immune Function. Cell Host Microbe. 2017;22: 757–765.e3.
- Hyun JC, Monk JM, Palsson BO. Comparative pangenomics: analysis of 12 microbial pathogen pangenomes reveals conserved global structures of genetic and functional diversity. BMC Genomics. 2022;23: 7.